


Though legally blind, Greg Bowman, PhD, can see with his peripheral vision and is able to read computer screens at high magnification. “One of the things that draws me to studying protein dynamics is that it’s something nobody can see,” said Greg Bowman, PhD, “so in some ways the playing field is a little leveled for me.”
When Greg Bowman was in second grade, he began to have trouble reading the board in his classroom. Playing goalie for his soccer team also became difficult. Over the following year, he lost most of his central vision due to an inherited genetic disorder, a form of juvenile macular degeneration.
“It gradually dawned on me that this would have a severe impact on my future life, with where I should live and work,” Bowman said. “But in retrospect, my parents handled it extremely well. I never felt like damaged goods or like I shouldn’t push myself to achieve as much as I could.”
Passionate about science, Bowman realized that experimental biology is not very accessible to those who are visually impaired. “Essentially, I see at low resolution, mostly with my peripheral vision,” he said. “I can navigate hallways and laboratories, but I can’t read the small dial on a pipette.”
So, Bowman found another route into the field: Computer science could be applied to biological problems and, with computers, he could zoom in to 16 times magnification.
Bowman, PhD, now an associate professor of biochemistry and molecular biophysics at the School of Medicine, is legally blind; he leads one of the largest crowdsourced computational biology projects in the world.
The effort is aimed at understanding how proteins — the raw materials that make up our bodies — fold into their proper shapes to keep our bodies running properly. Proteins are vital cellular machinery, and understanding how they assemble and function — or malfunction — could shed light on many of the most vexing problems in medical science.
The project is called Folding@home. It relies on the power of tens of thousands of home computers to perform the complex calculations required to simulate protein dynamics. Volunteers from all over the world install a program that runs those calculations when a computer otherwise would sit idle. Often motivated by personal interest, the participants get to select their area of contribution, whether it’s boosting cancer understanding, preventing Alzheimer’s disease or fighting antibiotic resistance, among others.
This map shows active Folding@home users at a single point in time.
With this networked computing power, Folding@home is, essentially, one of the world’s largest supercomputers.
“There are some traditional supercomputing folks who might take issue with that characterization,” Bowman said with a laugh. “Rather than a single massive machine, Folding@home is a distributed computing network. But in terms of raw computing power — the sheer number of calculations it can perform per second — it’s on par with the world’s biggest supercomputers.”
With Folding@home, Bowman and his colleagues are zooming in on proteins much more than 16 times. Indeed, they are getting as close as physically possible — down to the atomic level. Many important biological processes that proteins perform take place over milliseconds to a few seconds.
“To model just one millisecond of folding, even for an average-size protein, on a top-of-the-line MacBook Pro, it would take something like 500 years,” Bowman said. “But with Folding@home, we can split these problems into many independent chunks. We can send them to 1,000 people at the same time. Running those calculations in parallel, we can take these problems that would have taken 500 years and instead solve them in six months.”
Bowman got started on this work in the lab of Folding@home founder Vijay Pande, PhD, of Stanford University. After heading the project for 18 years, Pande chose Bowman, who completed a doctoral degree and postdoctoral research at Stanford, to take over leadership.
“Greg has a unique combination of skills,” Pande said. “He has the technical chops to lead this complex project, and he has the people skills to manage the distributed nature of it, especially the fact that it involves so many different kinds of people — scientists and nonscientists alike. Greg also has great vision for the future of this project.”
Folding@home’s massive computing capacity is crucial to understanding protein behavior, a problem Bowman calls a classic grand challenge in biochemistry and biophysics. To do its work, a protein must fold into its proper form. If it doesn’t, things go wrong.
Bowman understands this more than most.
A protein misfolds, then blindness
In childhood, Bowman’s vision loss was attributed to Stargardt disease, a form of juvenile macular degeneration caused when a protein that removes waste from retina cells doesn’t fold properly and can’t do its job. As a result, light-sensing cells in the retina become overwhelmed with waste and die, causing loss of central vision.
Later, with the advent of genetic testing, a basic microarray test did not find mutations in either of Bowman’s two copies of the gene that causes Stargardt’s disease — called ABCA4.

Greg Bowman’s team listens to a presentation at their lab meeting. The team is trying to unravel the role of protein dynamics in health and disease.
“As we’ve learned to classify diseases based on genetics rather than symptoms, my doctors backed off that diagnosis, no longer sure it was appropriate,” he said. “But it was an obvious candidate gene. Mutations in the protein ABCA4 are likely to interfere with its job pumping waste products out of retinal cells. We did sequencing at the time to look for mutations but didn’t find anything.”
More recently, sequencing technology has improved. New vision-related sequencing panels can identify mutations in what are called splice sites of the genes, which are difficult to analyze due to their repeating sequence patterns.
“In a new sequencing panel, three of the four mutations that popped up were in ABCA4,” Bowman said. “So, these are smoking guns, if you will. We are now looking into having sequencing done for my parents and my brother. Neither of our parents has any vision issues, but my brother and I do. We should be able to see what mutations we have in common and where we differ.”
Now that Bowman has a lead on what mutations are likely to have caused his own vision loss, he is interested in harnessing Folding@home to see what is going wrong with the protein and whether there is some route to fix it. Currently, there is no available structural information about ABCA4.
“We have found some related proteins that we hope to use as a template for trying to guess the structure of ABCA4, and then we can run simulations of both the normal protein and the mutated forms of the protein to see how they change,” he said. “Even if I can’t fix my own vision issue, it would be really cool to contribute to the fundamental base of knowledge that could help researchers do that for future people who come along with these genetic mutations.”
Studying at the atomic level
Because Bowman studies proteins at their most elemental level — looking at changes in individual atoms — his team can study a surprisingly wide array of biological problems. In addition to the early work on ABCA4, his lab also has well-established projects looking at antibiotic resistance in bacteria, proteins that contribute to Alzheimer’s disease and molecules involved in the extreme virulence of the Ebola virus.
“We’re in the nice position of having, essentially, a very rare microscope,” Bowman said. “Folding@home and all of the simulation tools and analysis tools that we’ve developed let us jump into a lot of different areas at once. We’re asking a lot of very basic science questions — how do things work in general, and those answers apply in many different contexts.”
Bowman envisions a future where Folding@home serves as a starting point for precision medicine, studying specific mutations in proteins and developing therapies to fix them. Right now, scientists often have only one structure of any given protein to study, if any structural information is available at all.
By watching Beta lactamase in motion, researchers have found “cryptic pockets,” weak points in the protein that could be targeted with drugs. Beta lactamase is a protein that some bacteria deploy to protect themselves from antibiotics.
Proteins largely have been studied using X-ray crystallography. In this technique, proteins are treated with chemicals, artificially freezing them into a particular shape, and then viewed by bouncing an X-ray beam off of them. However, this only gives static pictures of proteins in their “crystal” form. For example, beta lactamase, a protein that some bacteria deploy to protect themselves from antibiotics such as penicillin, has a well-documented, long-studied crystal structure. But that structure only represents a single snapshot of beta-lactamase at one moment in time.
Proteins often consist of hundreds or thousands of amino acids forming long chains that continuously bounce around in all directions.
“That snapshot contains valuable information,” Bowman said. “But it’s kind of like seeing a picture of a construction vehicle in a parking lot and trying to guess what it does. Really, what you would like is to watch this thing move around and see how it works together with other machinery to, say, build a building. We’re interested in watching how every atom in a protein moves — as it’s being assembled for the first time and as it goes about its jobs. And one genetic mutation changes maybe a dozen atoms out of thousands. We want to understand what that does to the entire protein.”
Fixing misfolds with new drugs
Among several projects, Bowman’s lab is using Folding@home to seek new drugs to combat antibiotic resistance. Watching the movement of beta lactamase, for example, already has revealed what Bowman calls “cryptic pockets,” weak points in the protein that could be targeted by drugs but that are not visible in the long-studied snapshot of this protein. The cryptic pockets only reveal themselves when the protein is moving.
Bowman also is using this rare microscope to study Alzheimer’s disease. A protein called APOE is responsible for transporting cholesterol through the body. The normal version of this protein is called APOE3. For reasons not yet known, a variant called APOE4 is associated with an increased risk of developing Alzheimer’s disease with age.
“Having one or two copies of APOE4 increases a person’s risk of developing Alzheimer’s anywhere from 3- to 15-fold,” Bowman said. “But APOE3 and APOE4 differ by only one amino acid — one protein building block out of 300 that make up the entire protein. We are trying to understand why one version of the protein is normal and the other is neurotoxic even though their structures are so similar. It’s a challenging problem to study because APOE is an extremely dynamic, floppy protein. It’s been hard to identify its structure using typical techniques, such as crystallography.
“If we can understand these structural differences, hypothetically, we can start developing small molecules that push neurotoxic APOE4, nudging it to look more like the neutral APOE3 variant,” he said. “And if we succeed in doing that, we might have a useful therapeutic. Rather than a typical ‘inhibitor’ drug, this would be a new type of compound that some people are calling a structure-corrector.”
Building on the discovery of cryptic pockets in beta lactamase, the researchers also are looking for weak points in Ebola virus, a deadly viral infection that causes vomiting, diarrhea and internal bleeding. On average, the disease is fatal in about half of all cases, with death rates ranging from 25 to 90% in past outbreaks.
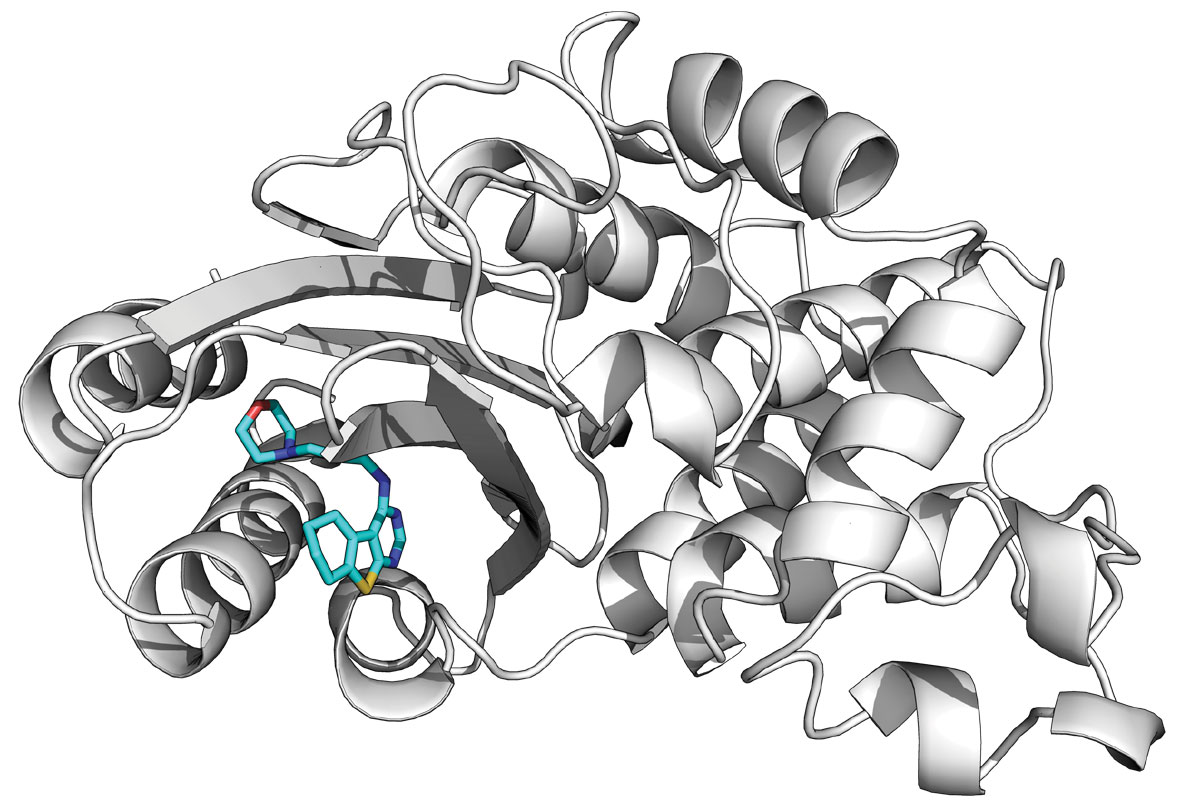
An example of a compound bond in a cryptic pocket.
Researchers are working to develop an Ebola vaccine or other therapies, but so far, the best treatment is intensive hospital care, including intravenous fluids and oxygen.
“Despite its virulence, Ebola has only seven proteins in its genome,” Bowman said. “These proteins interact with each other and with the molecules in our cells to hijack the infected person’s cellular machinery to reproduce the
virus and suppress the immune response.”
Conventional wisdom says these types of protein and RNA interactions can’t be targeted with conventional drugs because they don’t offer deep pockets where a small molecule inhibitor would fit to block their activity.
“If we could knock out some of these protein-protein interactions, how devastating would that be for the virus? That’s the kind of question we’re interested in,” Bowman said.
Nucleoprotein from Ebola virus in motion.
“I think this is a really exciting time,” Bowman added. “We’re starting to see some tangible outcomes that have the potential to impact how we think about diseases. We’ve already identified cryptic pockets and developed small molecule inhibitors for those pockets that interfere with both beta lactamase antibiotic resistance and with one of the protein-protein interactions that makes Ebola so deadly.”
As Bowman sees the world a bit differently than most, Folding@home offers scientists a different look at long-studied proteins, revealing solutions to biological problems that might otherwise remain hidden from view.
Julia Evangelou Strait is a senior medical sciences writer in Medical Public Affairs.

Published in the Autumn 2019 issue